Research
Our Mission
We are entering a new era of scientific discovery, where machine learning is transforming how we understand and engineer the physical world. This creates an opportunity to learn, predict, and reason about chemistry in entirely new ways. Our research combines machine learning and molecular simulation to study chemistry as it really happens - in dynamic, heterogeneous, and often messy environments. We pursue both new algorithms/methods and new chemical understanding.
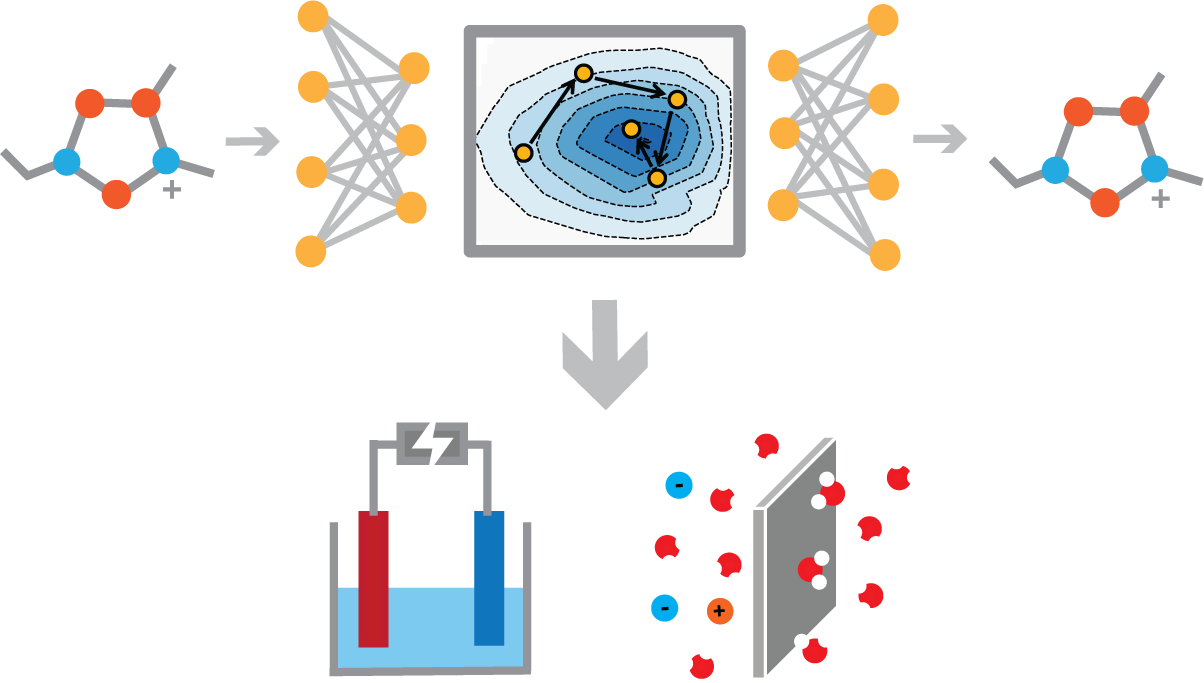
Physics-Aware Machine Learning
Machine learning models are now a routine workhorse for atomistic simulation and molecular design. The real test comes when these models are applied to complex chemical environments. Chemistry in the real world is messy and electronically complicated, and getting these systems right often requires more than just bigger models and more data. Can physical knowledge bridge this gap, and how should it be incorporated? Our group develops physics-aware machine learning algorithms and tools to tackle these challenges, including machine learning interatomic potentials (MLIPs), active learning strategies, and generative models for molecular simulation and design.
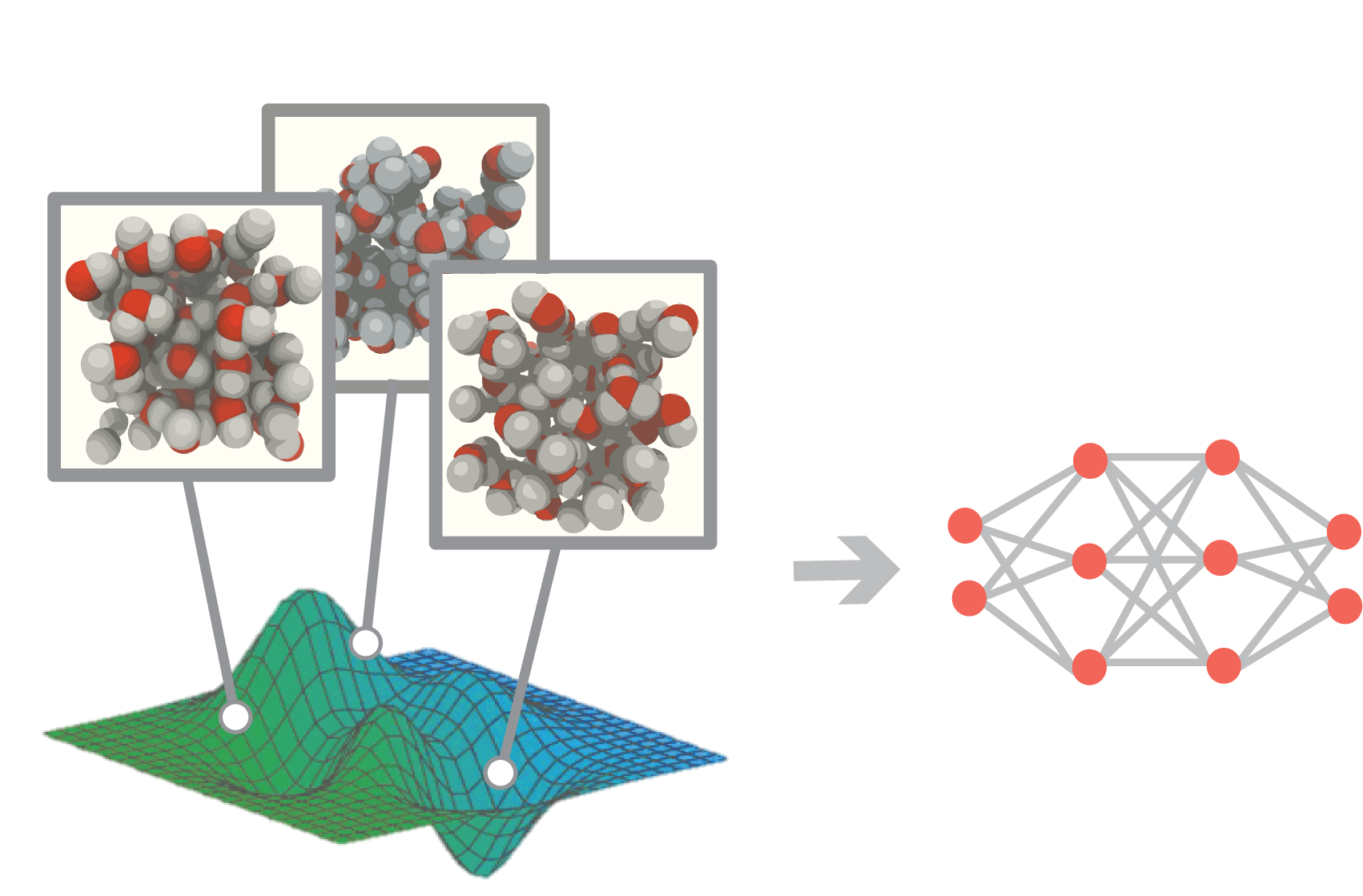
Learning What to Learn
Scientific discovery is becoming a conversation between researchers and intelligent algorithms. Every simulation (or experiment) teaches us something new, but figuring out what to learn next becomes increasingly difficult as data become more expensive to obtain and systems move beyond familiar regimes. Our group develops mathematically grounded methods that help decide what data to collect, where to explore next, and how to learn as efficiently as possible. These intelligent algorithms combine ideas from uncertainty quantification, active learning, and learning under distribution shift.
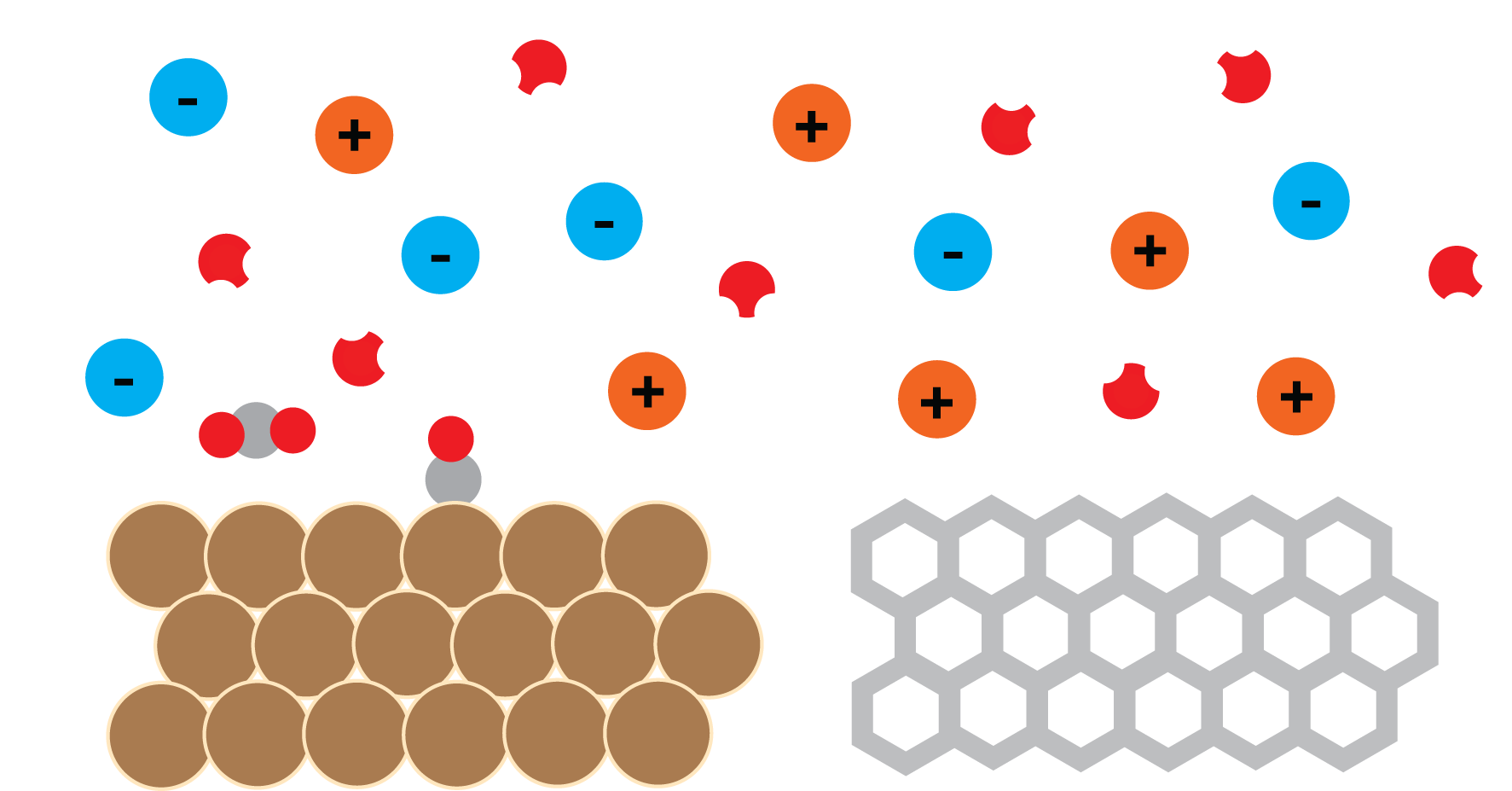
Engineering Functional Materials
Designing better materials can often feel like a game of whack-a-mole. Improving activity may reduce stability, increasing selectivity may decrease conversion, and small changes in composition can lead to dramatically different behavior. Our group studies the molecular and electronic origins of these tradeoffs and looks for opportunities to break them. We use these insights to guide the forward design of heterogeneous catalysts, 2D materials, and other functional materials for energy and sustainability applications.
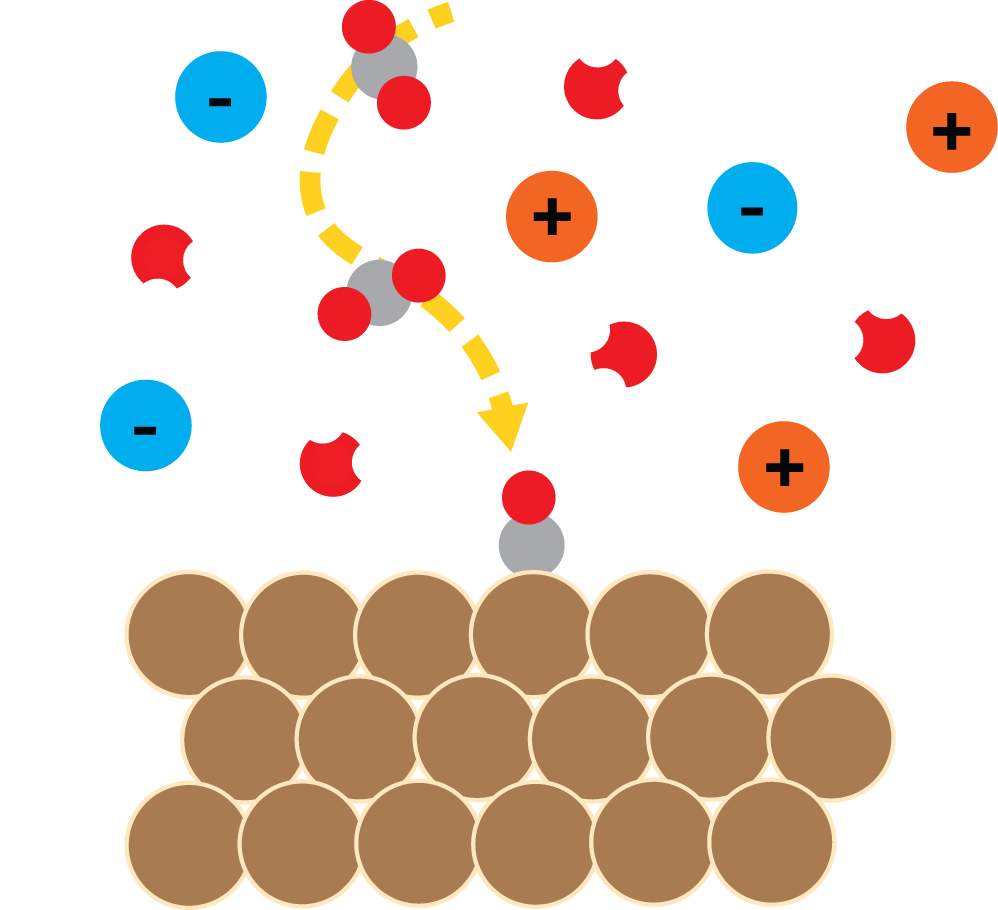
Engineering Interfaces and Solvents
Catalysts often get all the attention, but it is only half the story. The surrounding electrolyte can be just as much of a catalyst as the catalyst itself! The challenge is that these environments are dynamic, collective, and often difficult to characterize from experiments alone. Our group combines statistical mechanics and first principles theory to understand how solvents, electric fields, and fluctuations determine chemical behavior. We use these insights to engineer new electrolyte-design knobs for controlling catalysis, separations, and electrochemical processes.